Scott Carpenter, a systems administrator for a software development firm in Knoxville, TN, has been a bigfoot field researcher since he had an experience with one in 2009. His blog: https://bf-field-journal.blogspot.com documents his many searches for bigfoot in the Great Smokey Mountains National Park and surrounding public areas, where he has illegally placed hair traps, food stations, and stationary trail cameras without a Park permit. He has written two other books, The Nephilim Among Us: The Identity and Origin of the Sasquatch and Other Mysterious Creatures and The Bigfoot Field Journal, Volumes 1 and 2, the latter being a compendium of his bigfoot experiences, including those reported on his blog. Carpenter is perhaps best known for his many trail cam video stills of alleged bigfoots. His technique is to place the camera on his backpack facing rearward, and running continuously. Later he inspects the footage, frame by frame, looking for the slightest hint of anything remotely resembling a bigfoot or lately also a dogman. With his active imagination, not surprisingly, he finds what he is looking for among out-of-focus, blurry partial images, often obscured by foliage. He then tries to enhance the image with software called Blurity!, the manual for which specifically states that: “Blurity is for use on images that are properly exposed,” and “Blurity is not for severely focus-blurred images.” Neither requirement is usually met by his images. Finally, to convince his audience totally, Carpenter has an artist draw an image of what the subject might have actually looked like. What could possibly go wrong here? These many images and drawings are found in his blog. An example is found on pp. 123-124 in this book. From my experience, Blurity! can sometimes leave artifacts in the photo, especially where there is underexposure or no motion blur. Again from the manual: "Blurity is not for when some parts of an image are already sharp.”
Carpenter became involved in the Sasquatch Genome Project (SGP), http:/sasquatchgenomeproject.org, in 2011 and has submitted over 30 (his count) hair samples to “The Study”, 11 of which were chosen to be among the 111 samples accepted by the study. “The Study” resulted in the now famous, self-published paper (Ketchum et al., 2013), which is the subject of this, Carpenter’s latest book. He maintains he was an insider (pp. 6-7) and openly admits “…that Dr. Ketchum did not favor the publishing of this book. She preferred to ‘let sleeping dogs lie.’” (p. 7) From this review it is clear why; however, she continues to give sensationalist radio talk show interviews where she and her sympathetic interviewers control the narrative.
Throughout the book, Carpenter repeats the claim in the Ketchum et al. (2013) title and paper that “three whole genomes” were sequenced. Actually, the sequences of Samples 26, 31, and 140 were aligned with human Chromosome 11 only, which has approximately 135 million base pairs (Mbp). The human nuclear genome has approximately 3.3 billion base pairs in 22 pairs of chromosomes plus two sex chromosomes. Their Samples 26, 31, and 140 sequences only had 2.7 M (million)bp, 0.53 Mbp, and 2.1 Mbp respectively, small percentages of even the Chromosome 11 total of 135 Mbp., and only miniscule 0.08%, 0.02%, and 0.06% of the human genome, respectively. Thus, Carpenter perpetuates this basic fallacy about the Ketchum et al. Study.
Carpenter makes much of the Ketchum et al. claim that the three nuclear DNA sequences (Samples 26. 31, 140) “aligned with one another” and therefore “came from the same species” (pp. 46, 121, 159). However, simple BLAST® (Basic Local Alignment Search Tool, the database query software of the National Center for Biotechnology Information, NCBI), comparisons show that: Sample 31 has only an 8% query cover (overlap) with each of S26 and S140, and that S140 has a 62% query cover with S26. Furthermore, where they do overlap, the sequences do not align well enough to indicate that the same species is involved: only 91% for S140 vs. S26 and 95% for S31 vs. both S26 and S140. In general, homologous sequences from individuals of the same species should align better than 99%. These statistics prove that the nDNA sequences from S26, S31, and S140 do not align well as they should, being produced from the same reference sequence, and that they are not from the same species. It’s hard to imagine that at least one of the nine coauthors would not have done these comparisons.
All the above is easily explained when one considers that Ketchum et al. used a human reference sequence to sequence S26, S31, and S140, which guaranteed that only conserved genes would be sequenced and that the three different species might not have the same conserved genes with humans in all cases. Unless the species is definitely known, not presumed, to be closely related (at least in the same family), use of a specific reference sequence is inappropriate, interjecting bias and making interpretation of results more difficult (smaller differences when doing comparisons of conserved genes, e.g. 95% vs. 99%). The human S31 matched the database overall much better than S26 or S140 (Hart, 2016a and b), because it was human and was sequenced from a human reference sequence. The preferred method of sequencing would have been de novo, which does not employ a reference sequence.
In addition to the fact that the three nDNA sequences do not align with each other, Hart (2016a and b) has shown that, in fact, they align very well with NCBI database (GenBank) sequences and other published sequences from black bear (S26), human (S31), and dog (S140). My peer reviewers agreed. Carpenter has never challenged these published, peer reviewed findings on anything like a scientific basis. This book would have been the ideal place to do so, but instead he maintains (p. 121) that “The genomes did not match any organism in the GenBank Database. The three genomes were from a completely unknown species….” without any detailed alignment results from the Ketchum et al. (2013) paper or his own research. Ketchum et al. never supported these claims with BLAST® search results, as Hart did for his results (2016a and b).
Another of the Ketchum et al. false conclusions pushed by Carpenter is “The data indicates (sic) that the Sasquatch …possesses nuclear DNA that is a structural mosaic consisting of human and novel non-human DNA." (pp. 110 and 271) Hart (2016b) shows this to be false in Figures 3 (Sample 26) and 4 (Sample 140). The figures were produced from thousands of GenBank sequences that matched Samples 26 and 140, respectively, better than 95%, most bear and dog sequences better than 99%. Human and primate matching sequences are very few in number and far between in these figures, never matching better than bear and dog, respectively, over the entire Ketchum et al. nDNA sequences.
It is not surprising that:
“The nuDNA sequencing and alignment was performed by the University of Texas at Arlington, Wakeland Laboratory. The Wakeland Laboratory Director refused to allow the Bioinformaticist to be listed as an author on the manuscript even though he wrote the whole genome section and the whole genome/bioinformatics materials and methods contained section in the Sasquatch Genome Study.” (p. 101)
Some of the conflicting results that I found with specific gene sequencing were dismissed by Carpenter as negative evidence disallowed by JAMEZ reviewers (p. 74):
“• The nuDNA did not contain the human TYR gene. This gene is associated with skin and skin pigmentation in humans.
• The nuDNA did not contain the human HAR1 gene. This gene is a “human accelerated region” that is associated with human neurological development.”
Specific gene sequences for some samples are found in Ketchum et al. Supplementary Table 3 and on the SGP website. [Note: Hairs plucked from Sample 26 are referred to as Sample 25 on the SGP website. Here both are combined as S26 for simplicity.] Simple BLAST® searches found the following matches in GenBank, which are discussed in my Paper 3 on my blog, https://bigfootclaims.blogspot.com/p/paper-3.html :
For HAR1: S26 polar bear from SGP website (surrogate, limited black bear data available in GenBank) The HAR1 result required searching the Reference Sequence GenBank database, which has whole genomes. At the time the “nucleotide” database had relatively little black bear data and no polar bear data. Ketchum et al. only searched the “nucleotide” database, so they came up with nothing and called it “unknown.”
TYR was never discussed in the paper or on the SGP website.
Other mixed results were found:
For Amel X: S26 unknown from paper Sup. Table 3 and dog (99.46%) from SGP website (Justin Smeja’s perhaps, as it accompanied him on the S26 sample recovery).
For Amel Y Exon 2: S26 polar bear 100%ID from SGP website (surrogate, limited black bear data available in GenBank). Ketchum found European polecat (Mustela putorius ) with only 83%ID, the result of only searching the nucleotide database (SGP Website Supplemental Raw Data). She said it meant that the actual species was unknown. Hardly, it’s a bear.
For TAP1: S26, S35, and 39b human; S10 dog; S33 human mtDNA, NOT nDNA; S43 and S44 unknown. Thus the TAP1 results in Ketchum et al. Table 7 are wrong.
Such mixed results cannot come from a single species. They are the result of contamination and different sample species, not "some sort of hybrid" as concluded by Dr. David Swenson (see below).
Says Carpenter (p.74):
“The gravity of these “negative” results become (sic) extremely important when considering the questions of contamination, identification and uniqueness of the subject tested.” (p. 74)
We couldn’t agree more.
On behalf of the SGP, Ketchum coauthor Aliece Watts attempted to submit a 669 bp mtDNA sequence under the name “unknown hominid” then later Homo sapiens to GenBank, which refused it. (p. 74 and Exhibit 15). The sequence covers the HV1 region and differs from all mtDNA sequences in Ketchum et al. (2013) by 3-10 base pairs (mutations). The sequence is not in the published study. It does not match the sequence in the SGP video on contamination (see below) either. So where did it come from? It matches 29 Homo sapiens sequences in GenBank with two mutations (99.70%) and no other hominins better than 93.12% for Homo heidelbergensis and 88.67% for chimpanzee (Pan troglodytes). It is a human or near human sequence.
One of Carpenter’s conspiracy theories is that, fearing prosecution for game law violations or even murder, Justin Smeja along with Bart Cutino and Tyler Huggins, submitted two known black bear samples to separate forensic laboratories, so that Sample 26, from which they would claim origin, would be determined to be a bear, not a human or nongame mammal. (pp. 132-133) Smeja, Huggins, and Cutino all deny this claim. (Exhibits 31 and 32, pp. 304-309)
Justin Smeja submitted to a polygraph test and passed all questions with “NO DECEPTION INDICATED.” (Exhibit 4, p.245). Two questions bear on the above:
“12) Do you truly believe that the piece of flesh you have processed, cut into pieces, and sent to various lab, is a piece of flesh from the unidentified animal you claimed to have shot from the "Sierra Kills" incident?
Answer: Yes Test Result: Passed NO DECEPTION INDICATED
13) Is the piece of flesh given to Wally [Redacted] from the same source as the piece of flesh you were instructed to send to a [redacted] lab by Bart Curtino?
Answer: Yes Test Result: Passed NO DECEPTION INDICATED”
Polygraphs tests are overall 87% reliable. Smeja passed 40 questions according to the analyst. (Exhibit 5, p. 248)
A key point made by Carpenter is that the three samples (S26, Cutino’s and Huggins’) all had different human mtDNA haplogroups, H1a, T2, and A respectively, and so could not all be cut from the same original stock sample collected by Smeja and sent to Ketchum. Actually, this is a false conclusion, as different people could have contacted the macro sample in different places. Carpenter only exhibits the first pages of the Cutino (p. 277) and Huggins (p. 278) reports. The full reports are found here:
https://bigfootclaims.blogspot.com/p/fi-na-l-r-e-p-o-rt-o-n-an-l-y-sis-o-f-s.html
https://bigfootclaims.blogspot.com/p/huggins-figures-continued.html
https://bigfootclaims.blogspot.com/p/bart-cutino-report.html
In the first place, Ketchum et al.’s S26 haplogroup determination H1a is not unique, H5e being equally likely. [Note: Throughout their respective publications both Ketchum et al. and Carpenter incorrectly use the term haplotype for haplogroup. Wikipedia explains the difference: https://en.wikipedia.org/wiki/Haplotype.] Family Tree DNA determined that Sample 26 had 16 extra mutations from H1a, so many that I found that a human haplogroup cannot be uniquely determined. Statistically, it has only one chance in approximately 224 million of being human, as determined by Poisson statistics (my blog, Paper 2). Most humans have only two to four extra mutations from any haplogroup. Eight of 18 full mtDNA sequences in the Ketchum et al. study had between 7 and 17 extra mutations, and have less than a 1% chance of actually being human, some much, much less. So what are they? Most likely, these samples are contaminated by one or more humans. It is also possible that some of these samples, except S26, are actually from mutant humans or a sasquatch much closer to human (and more distant from chimpanzee) than many have supposed. A key technical detail is that Family Tree DNA, which sequenced the mtDNA is a company that only does known human DNA analyses, using human specific primers, which means that any other species present, even the major species, would not be detected. I tested this hypothesis by sending horse, dog, and cat buccal swab samples to Family Tree DNA, and they returned the same result for each: failed to amplify. So a trace amount of human mtDNA would be amplified and sequenced just as if it were the only species present in an otherwise animal sample.
Secondly, further into the Huggins report it is stated that the human portion of his sample is identical to GenBank accession JQ705199, which is haplogroup T2b as proven by me (BLAST® alignment). The Cutino sample has an identical HV1 mtDNA sequence (BLAST® alignment), so it is also T2b. The authors of the Huggins report were careful not to mention the haplogroup explicitly, but from their description of JQ705199 it is clearly T, T2, or T2b, and definitely not haplogroup A:
“Further analysis indicates that it is a European haplotype; it occurs with highest frequency in East Europe (11%) and Caucasus (10%) (i.e. it initially originated from near the Caucasus mountains regions between the Black and Caspian Seas). It is not known to have originally occurred in East Asia, Southeast Asia, Australia, Oceania (i.e. New Zealand, Papua New Guinea, etc.), North America, South America or Central America.” (Emphasis mine)
“Haplogroup A” was an unfortunate place holder designation in a Huggins Report table (poor scientific reporting), indicating that the control from submitter Justin Smeja and the Huggins samples had identical haplogroups, not haplogroup A. Haplogroup A is a Native American haplogroup with origins exactly where the above description of T2 is said not to have originated.
Also, I found that both Cutino’s and Huggins HV1 sequences differed from four different haplogroup A accessions (MH553890.1, MH553825.1, MH553811.1, MH553742.1) in GenBank by the same nine topological mutations. Conclusively, neither sample is haplogroup A.
Importantly, the control samples from Justin Smeja matched exactly the two hide samples (frozen and dried) in the human HV1 region and in 13 of 13 human STR (short tandem repeats) alleles which amplified in one or both subsamples. Therefore, both Cutino and Huggins’ samples contained Justin Smeja’s contamination with very high probability.
Finally, nine of the sixteen extra mutations (73G, 709A, 930A, 3010G (a reversal), 4917G, C15452a, 15607G, 16126C, and 16296T) from H1a in the Ketchum Sample 26 are found in T2b, and a tenth mutation (16187C) defines haplogroup T2b3e. So contamination by Justin Smeja is likely present in Ketchum et al.’s Sample 26 too.
Incidentally, the great majority of the DNA in Cutino and Huggins’ samples was from black bear, as proven by cytochrome b sequencing and STR analysis with black bear primers, but then so was Ketchum et al.’s S26 a black bear, as proven by Hart (2016a and b) from the Ketchum et al. nDNA sequence (see above) and Sykes (2014) from a hair (see below).
Carpenter discusses human STR alleles on pp. 133-134, comparing one split Cutino sample (frozen) to a Ketchum et al. hair Sample 25 (from the Sample 26) in their Table 5. His table on p.134 does not show the Cutino dried sample, which had more alleles, or the Smeja control sample, which had even more (see above). Although the Ketchum et al. and Cutino results did not agree in this context, it is clear from above haplogroup results, there are more than one human-like contamination in Sample 26.
Even proof of a common contamination, Justin Smeja, is not proof of a common origin since Smeja is known to have contacted all three samples. Early on I suggested to Melba Ketchum that she use black bear primers on her remaining Sample 26 to attempt to reproduce the results in the Cutino and Huggins Reports. This would have clinched the deal: either they would fail to amplify, in which case her S26 is not from a bear, or they would yield bear sequences and possibly the same bear sequences and alleles found by Cutino and Huggins. She did not respond to my suggestion. Instead, she destroyed her sample and urged Smeja to do the same.
Both Carpenter and Ketchum et al. claim in their respective publications that all 111 study samples were sequenced in the cytochrome b and HV1 regions of mtDNA with universal and mammalian primers, respectively, and that they matched human 100%. It is highly unlikely that all 111 samples field samples would be human by mtDNA. Such an effort to sequence 222 samples (2 x 111) would have easily cost $300,000 or more at market rates. No primer or sample sequences were ever published, however. The very same Sample 26 cyt b region was sequenced in the Tyler Huggins Report (see above) and found to be from a black bear. Show us your sequences please, Dr. Ketchum, just as Huggins did (and which I confirmed was from a black bear).
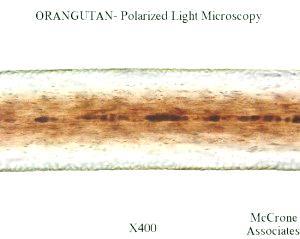
Microscopic hair analysis is discussed in Chapter 10, p 109, where Carpenter shows a comparison of human and “sasquatch” hair, similar to the Ketchum et al. (2013) Fig. 5. What is called “sasquatch hair” for Sample 26 is clearly black bear hair, with the telltale continuous, nodose, corpuscular medulla of flattened, disc shaped “cells”. Also the medullary ratio (MR = diameter of medulla/diameter of whole shaft) is in the range of black bears (usually 0.25-0.5) and different from what other researchers have proposed for sasquatch, i.e. thinner, hollow or no medulla (Meldrum, 2006, 2013). The great apes, including humans, have very thin (MR less than 0.2, often much less) and discontinuous, fragmental medulla, hollow medulla, or no medulla as in Carpenter’s and Ketchum’s human photos and many others. Ketchum et al. stated in their paper that “Most of the submitted hairs were not microscopically consistent with any of the hairs from the reference collection of common animal hairs that included human, cat, dog, cow, horse, deer, elk, antelope, moose, sheep, fox, bear, coyote, wolf, rat, mouse, monkey, beaver, squirrel, llama and others.” However, they misidentified S26 hairs and maybe others as well. Other photomicrographs of black bear hair which match extremely well the Ketchum et al. (Fig. 5), Carpenter (p. 109) figure, and black bear photomicrographs in my private collection, can be found at:
https://alaskafurid.wordpress.com/2009/11/02/bear-black/
https://archives.fbi.gov/archives/about-us/lab/forensic-science-communications/fsc/july2004/research/2004_03_research02.htm
http://www.nsrl.ttu.edu/publications/specpubs/sps/SP55.pdf
http://people.cst.cmich.edu/swans1bj/hair%20id/ursus/ursus.html
“The Electron Microscopy was performed by the University of North Carolina, Chapel Hill, North Carolina. The director was unhappy to be part of the blind study, refusing to allow his facility to be associated with the Sasquatch Genome Study. He did, however, give permission for the data/photographs to be used in the Sasquatch Genome Project.
The University of North Carolina, Chapel Hill Electron Microscopy results were confirmed by Texas A&M University, College Station, TX. Texas A&M was paid for their work.” (p. 101)
As mentioned by Carpenter (p. 117), electron microscopy results were unusual, showing sections of single-strand DNA alternating with normal double-strand DNA. Only viruses are known to have single-strand DNA. I asked coauthor Dr. Andreas K. Holzenburg, the TAMU electron microscopist, what could be the causes of this phenomenon (such as microbial, thermal, chemical or photo degradation, or sample preparation), and he replied:
“My involvement was limited to evaluating electron micrographs of DNA. This evaluation by itself does not permit to lay claims to the existence of paranormal phenomena - so please direct inquiries directly to Dr. Ketchum.”
I expected a more enlightening response from the author of the electron microscopy section of the Ketchum et al. paper.
Carpenter’s interpretation is that:
“The single stranded gaps showed that parts of the genome did not align. This will happen with hybridization since the DNA sequences are different for different species and will not line up identically as with a single species mating.” (Emphasis his)
Carpenter does not understand the process of fertilization. The sperm and the egg each contribute whole chromosomes, not single strands of DNA. Then there is exchange of whole genes between like chromosomes.
It is doubtful that a double-single-double DNA strand could replicate in mitosis; there are no known examples. One complementary strand mRNA would terminate at a single-strand juncture, leading to only one viable daughter instead of two. Single strand DNA is very likely the result of post mortem degradation, not some new form of life as implied (but certainly not proven) by Carpenter and Ketchum et al.(2013).
As part of his conspiracy theory, Carpenter asserts (p. 181) that, while I (Haskell Hart) claimed that:
“I also reached out to the Ketchum coauthors, asking for their comments on a previous version of my papers. I also got no response from any of them.”
and that I did not respond to his (Carpenter’s) request that I provide him with:
“• Which co-authors did you contact?
• What method did you use to contact them (email, phone, etc.)?
• What was the exact response of each one (denial, no- response, etc.)?” (p. 181)
However, I never received such a request from Carpenter via US mail, email, telephone conversation, or any other means. So, Mr. Carpenter, What method did you use to contact me (email, phone, etc.)? And, did you ever contact the coauthors to determine if they ever received my emails? If I had received such a request, I would have sent Carpenter the emails from me to Ketchum coauthors, all of which went unanswered. For example:
“Apr 18, 2013 at 4:38 PM
Dr. Prychitko,
Attached is my review of "Novel North American Hominins...", which you coauthored, with supporting documents.
The paper contains references to some missing supplementary figures and data and has some unsupported conclusions.
I sincerely hope that an addendum will be published to clarify these and other matters outlined in the review so that this important research can be presented in the best possible light.
Haskell Hart”
Identical letters were also sent to coauthors Holzenburg, Watts, and Wojtkiewicz, the only ones for which I could find an email address.
Later I sent the following email to these same coauthors:
“Jun 19, 2014 at 8:39 AM
Dr. Prychitko,
Attached is a paper I wrote on the Ketchum DNA paper which you coauthored. You should be interested. Comments most welcome.
Haskell Hart”
Again, no answers were received. I also sent a final version of my paper (Hart, 1916a) to coauthor Fan Zhang and to Melba Ketchum, requesting comments. Fan did not respond, and Ketchum responded with unpublished results from an anonymous consultant, who did not disprove my claims, rather verified my 95%ID match of S26 nDNA to human the only species he bothered to compare it to. (I matched it to black bear 99+% at the time, 100% more recently).
Carpenter claimed (p. 182):
“With the stigma and possible dire consequences associated with the Sasquatch Genome Study, it would be no surprise that if contacted by Meldrum or Hart, the co-authors of the Study would not respond to high profile vocal critics of the Sasquatch Genome Project.”
So Carpenter says I did not contact coauthors, then immediately says they would not have responded anyway. This is a red herring and a convenient cover for the lack of response from coauthors, which belies the fact that the coauthors’ names and their affiliations are clearly present on the first page of the Ketchum et al. (2013) paper antwaty. The coauthors were apparently under nondisclosure agreements (NDAs), and could not legally respond if they wanted to. Carpenter either did not ask Ketchum coauthors if they had heard from me, or if he did contact them, he did not say so.
Carpenter closes Chapter 13 with the following comment:
“In my opinion, it is evident, when one closely examines all the different criticisms of the Sasquatch Genome Study, none have any foundation in fact. The criticisms are merely 'journalistic clutter' used to confuse, frustrate and complicate the matter."
Carpenter has refused to recognize, “closely examine,” or respond to any scientific criticism of the Ketchum et al. (2013) paper, for example those in my blog https:/bigfootclaims.blogspot.com, and publications (Hart, 2016a and b). I even wrote him an email with my preliminary results, asking for his comments, but I received no response:
“Oct 19, 2013
Mr. Carpenter,
In the interest of open-mindedness and honesty I invite you to examine closely the Ketchum Supplementary Figures 5 and 6, by ZOOMING IN and PANNING (otherwise they are unreadable). I attach them for your convenience. You will see that Supp. Fig. 5 is a phylogenic tree with a chicken (Gallus gallus), a mouse (Mus musculus), and 29 species of FISH, all supposedly related to a sasquatch. See attached list of the common names of these fish. Supp. Fig. 6 shows a tree with the mouse (Mus musculus). These indicate that the BLAST searches from which they derive are flawed or misinterpreted, as my paper points out….”
Again, there was no response from Carpenter. He refuses to acknowledge my scientific critique of the Ketchum et al. paper.
Carpenter devotes Chapters 8 and 9 to peer reviews of the Ketchum et al. (2013) paper, a key element in his theory of conspiracy of establishment science against anything new and revolutionary. My in depth analysis of every reviewer comment can be found at:
https://bigfootclaims.blogspot.com/search?q=Peer+reviews
To summarize: There were four Nature reviewers, who went through one revision before rejecting the paper, and two JAMEZ reviewers, who accepted the second version of the paper “with revisions”, which were never made. Melba Ketchum subsequently bought the journal and self-published the paper on the DeNovo journal website and the Sasquatch Genome Project (SGP) website.
Reviewer comments below are in order of decreasing reviewer consensus. Bold numbers in brackets [..] are the number of reviewers (of the six total) who concurred. From my blog:
(1) Inadequately substantiated thesis. Results do not support conclusions. [6]
(2) Results of genomic analysis superficially treated. Results are not adequately documented. More stepwise in depth treatment needed. Need more information on analysis of whole genomes. Analytical credibility could be improved by fully leveraging information from next generation sequencing. Sequences should be available. Bioinformatics should include reference sequences from expected contaminants. [4]
(3) Hominin not proven. Primate a better term. [4]
(4) Phylogenetic trees not clear, inadequate, or give mixed results. [4]
(5) Concern about monitoring submitter contamination. Results indicate contamination. [4] Did avoid contamination. [1]
(6) S26 provenance murky at best. [2]
(7) Selectively aggregated sequences to support a favorable placement among primates. Reference sequence bias in developing consensus sequence. [2]
(8) Poor agreement between mtDNA and nDNA results across samples. S31 does not align with S26 and S140. [2]
(9) Change "unknown" hair morphology to "novel." Should cross reference human morphology. Expected mistakes in hair identification made. Needs statistical analysis. [2]
(10) Quality control is difficult with contract labs. [1]
(11) S26 - ethics of shooting. [1]
(12) Q30 scores important. Seemed to justify publication. [1]
(13) Need better photographic evidence. Stick structures inappropriate. [1]
(14) mtDNA is all "poorly" human. Would expect some other lineages. [1]
(15) Unknown sequences more likely from unknown microorganism. [1]
(16) Electron microscopy suggests DNA damage. [1]
You will note that, although I was not aware of these peer reviews until after I had published my major criticisms in my blog, virtually all of the reviewer criticisms were the same as mine. See above for these, also my blog.
More conspiracy claims as Carpenter slams the peer review process:
“In the modern era the ‘Peer Review’ process has been corrupted, it is now used by ‘gate keepers’ to exclude evidence that goes against the accepted dogma of the discipline. It is no longer a method to bring new discoveries to the scientific community. It now protects the ‘status quo’ allowing just a small minority to control the ‘version of the truth’ they want disseminated. This was definitely evident in the Sasquatch Genome Study, however there are other examples of this suppression of the truth.” (p. 65)
Nothing could be farther from the truth. The peer review process prevents the proliferation of unimportant, poorly written, poorly documented papers with deficient experimental protocol, poor execution, questionable results, and unsupported conclusions from cluttering up the literature. There are other means besides professional journals to publish these kinds of research, for example self-publishing as did Ketchum et al. The peer review process ensures that the major journals are affordable (length is important) and worth subscribing to by individuals and libraries alike. The process, though not perfect, is honored by all reputable scientists, most of whom have had papers rejected, and virtually all of whom have been called upon to review the work of colleagues, a duty which takes time away from more rewarding activities. I have been on both ends of this process, and I even contributed lengthy calculations to verify an author’s results in one case, which was acknowledged gratefully in the final published paper. It’s too easy for nonscientists like Carpenter and Robert Lindsay (p. 66-67) to criticize something they do not fully understand and have not been involved in, especially when the process does not support their agendas.
Carpenter is harsh in his treatment of Oxford Professor Bryan Sykes and his independent DNA study of purported sasquatch samples (Sykes, 2014): “Chapter 12 – Dr. Sykes, A Wolf in Sheep’s Clothing.” (p. 137) His major claims were that Sykes colluded with the editors of Nature to delay the peer reviews of Ketchum et al. (2013) so he could beat them to the punch with his own study, and that Sykes cherry picked the samples and/or results in his paper (2014) to ensure that no sasquatch DNA was found. Neither claim seems likely, especially that Sykes would not want to have found DNA evidence of sasquatch, even a year after Ketchum thought that she did and he had proved that she didn't for Sample 26.
Though there is disagreement on whether the study received 57 or 95 hair samples, 37 were selected “based on their provenance or historical interest,” of which 30 amplified and produced sequences in the 12S rRNA region of their mtDNA. The results were all known animals, and I confirmed the results with BLAST® searches of GenBank. One was human, which Sykes confirmed with a separate HV1 sequence that matched the human reference rCRS exactly, but not that of Neanderthal Man, Heidelberg Man, or Denisovan Man.
Very unfortunately, Sykes did not indicate the “provenance or historical interest” of each of these 30 samples (only the country or US state) in his Table 1 of results (Sykes, 2014). Had he done so, especially by associating well-known contributors with their samples, the cherry picking claim might not have held water. Sample submitters, some quite well-known, were acknowledged en masse at the end of the article. These were: Reinhold Messner, Peter Byrne, Justin Smeja, Bart Cutino, Derek Randles, Dan Shirley, Garland Fields, Loren Coleman, Betty Klopp, Marcel Cagey, Sam Cagey, Lori Simmons, Adam Davies, Dr Mike Amaranthus, Mike Long, Patrick Spell, Maxwell David, Mark McClurkan, Rob Kryder, Jack Barnes, Jeff Anderson, David Ellis, Steve Mattice, Brenda Harris, Stuart Fleming, Igor Burtsev, Dmitri Pirkulov, Michael Trachtengerts and Dmitri Bayanov.
At any rate, the cost of analyzing the 37 samples would be about $50,000 at the rate confirmed by coauthor Dr. Terry Melton, who supervised the analyses at Mitotyping Technologies, her lab in Pennsylvania. Chris Kummer, of the blog Die Tiefe, has located remaining samples at the Museum of Zoology, Lausanne, Switzerland. Those with the means and desire to verify results or produce new results from previously unanalyzed samples should contact Sykes coauthor Michel Sartori there.
Carpenter makes two false technical claims in this chapter: (1) that Ketchum et al. used a very similar cleaning procedure as Sykes, and so their hair analyses are as free of contamination as his were, and (2) that only hairs with a root have enough mtDNA to analyze. Actually, Melton describes her cleaning procedure in a reference (undoubtedly not read by Carpenter), and it involves ultrasonication of the hair in water. Ketchum did not do this, which may have made all the difference, in some cases. In addition, Melton also reports amplifying and sequencing DNA from hairs, some of which were less than half a centimeter in length, and some of which were decades old. So there is plenty of mtDNA in most hairs, even without a root.
Matt Moneymaker, quoted by Carpenter, says “The Sykes study is meaningless scientifically.” (p. 144). Not quite, Matt. Sykes did prove that:
1. It is possible to extract, amplify, and sequence mtDNA from limited amounts of aged animal hair samples,
2. The very limited 104 base 12S rRNA mtDNA sequence used was sufficient to identify all 30 sample species except those known to be similar: dog/wolf/coyote, white-tailed/mule deer, brown/polar bear (including hybrids), and the sheep/Himalayan tahr ambiguity discovered by me. There was no question, however, that none of the 30 samples, except the human one, was a primate.
3. Human contamination on hair can be eliminated by special treatment, including ultrasonication,
4. In spite of their preeminence and presumed interest in having their samples identify with sasquatch, sample submitters were only able to produce hairs from common animals, whose DNA matched that in GenBank,
5. Once again, Ketchum et al. Sample 26 is a black bear. That makes three independent laboratory DNA analyses plus my analysis of Ketchum et al’s own S26 nDNA sequence, and
6. As suggested by one of my reviewers, unnecessary expense can be avoided by screening hair samples by light microscopy before doing a DNA analysis. However, Sykes wanted to prove that his sample preparation and analytical technique were sound, regardless of the sample origin.
Note: Primate hairs are very different from those of the animals in Sykes’ study. Identification of hairs can be subjective and requires a large collection of reference samples or photomicrographs and, in some cases, statistics of reference and unknown populations - any two compared samples may not be representative of their species. Difficult identifications, especially from among species from the same genus, can require considerable experience, or may remain ambiguous at the level of speciation.
In Chapter 15 Carpenter makes much of the supposed expert opinion of Dr. David Swenson, a biochemist with 38 publications in the field of oncology. Swenson was one of the first genuine scientists to jump on the Ketchum bandwagon with an armchair opinion (based on a cursory examination without knowing or understanding the details), which was published by Ketchum on Facebook and excerpted below. From examination of his otherwise impressive publication list (Exhibit 23, p. 289), extolled from the mountain tops by Melba Ketchum, it is very clear that Dr. Swenson had no professional experience in forensic wildlife DNA analysis or anything like it. He is not a geneticist either. Yet he commented on February, 17, 2013 on Melba Ketchum’s Facebook page (p.198):
“My desktop had difficulty with a blast analysis of the consensus sequences….My opinion of the creature is that it is a hybrid of a human mother and an unknown hominid male, just as reported…. Sasquatch is real, as proven by genetic analysis”
Sir, you are not entitled to a real scientific opinion on this subject if you cannot do a BLAST® analysis to check the conclusions of Ketchum et al. Having seen this quotation in early 2013, I sent Dr. Swenson by Facebook messaging, a BLAST® tip (plain to see in their help page) to resolve his difficulty. He did not respond, but shortly thereafter he closed his FB page down or blocked me from seeing it, and began posting his own BLAST® results.
Later on he is starting to see the light (p. 201): “There are random good matches for some sequences from various species such as dog and panda…” (precisely my findings). Unfortunately, he wasn’t smart enough to realize that his findings were not "random," and to pursue the panda clue farther to see that there are even better matches to polar bear and black bear for Sample 26. Sample 140 is from a dog, as discussed above.
Finally, on the Joe Rogan show (p. 200), Dr. Swenson admits his legitimate doubts, but never admits his significant change of opinion.
“(It) Could be a real animal.” (Emphasis mine)
“… as a scientist, I want the damn thing to bite my fingers off before I am going to believe it.” (Emphasis mine, quite a switch from his early armchair opinion)
Joe Rogan: "Do you believe Sasquatches exist?"
Swenson: "Well, I would not go this far. Let’s put it this way. One thing I know for sure is there is an unknown animal in the Pacific Northwest. Let’s just leave it like that."
Obviously, Swenson is confused and/or conflicted, but Carpenter blithely attributes this hedging to a fear of a stigma attached to sasquatch in the scientific community, completely ignoring his own quotation of Swenson (p. 201):
“I can say what I choose to, and will speak truth to power. I don't give a damn what others think.”
[Note: Swenson is retired. So am I.]
Another of Carpenter’s so called supporting scientists, Alister John Marsh, has made the claim that Ketchum Samples 1, 12, 36, 39b all haplogroup T2b, have in common five unusual mutations not found in normal human T2b. (p. 194 ff and Exhibit 27, p. 296) Specifically, he claimed 146T, 16187C, and 16189T were absent in bigfoot samples but always present in normal human T2 and T2b. Also, he claimed that 73G and 263G were present in bigfoot T2 and T2b, but absent in normal human T2 and T2b. As I pointed out to Marsh in early 2013 on Ketchum’s Facebook page, his reference for the human samples was RSRS (Reconstructed Sapiens Reference Sequence), whereas Ketchum’s for bigfoot was rCRS (reconstructed Cambridge Reference Sequence). These reference mutations differ by exactly these five mutations above, among others, http://phylotree.org/resources/RSRS_vs_rCRS.htm. The T2 mtDNA Project on Family Tree DNA demonstrates this clearly. Of 233 T2b’s in the T2 Project only seven had 16189C, otherwise all agreed with T2b’s from both Ketchum et al. AND GenBank. https://www.familytreedna.com/public/T2?iframe=mtresults. Ketchum’s bigfoot results are rCRS; Marsh neglected to select this option on the T2 mtDNA Project webpage, instead getting the RSRS option for mutations by default. Prof. Todd Disotell has a phrase for this: “Rookie error.”
I also did BLAST® comparisons of The Ketchum T2b samples against 98 GenBank sequences with T2b in their titles, with the following results: all 98 had 73G, 97 of 98 had 263G, 92 of 98 had 146T, and all had 16187C and 16189T. Bigfoots have the same 73G and 263G mutations as database T2 and T2b”s; I aligned them with rCRS. But neither bigfoots nor normal T2b’s have “mutations” 146T, 16187C, or 16189T, because these bases are the same as in rCRS (but not in RSRS), except 39b had a 16189C mutation. The complete Cro-Magnon mtDNA genome (Genbank KC521456.1) mentioned by Marsh (p. 297) is T2b as confirmed by me, and it also matched Ketchum et al. and virtually all GenBank T2b’s (see above) at 16187C and 16189T (over HV1) and at 146T, 73G, and 263G (HV2 region). Hence Marsh’s claim that Cro-Magnon matched Ketchum et al. but not GenBank T2b’s is false (p.196), again due to reference sequence errors. Cro-Magnon Man is a modern human genetically. Hence, Marsh’s theories of the evolution of bigfoot mtDNA are all falsely based. (Exhibit 27, p. 296 ff.) He’s a genealogist, not a geneticist.
By the way, Ketchum deleted my related 2013 comment to Marsh on her Facebook page, and Marsh promptly sent me an email requesting that I refrain from any more public comments on the subject until he checked his results. He never responded to my subsequent inquiry on the results of his check, but he has admitted his error elsewhere. Carpenter should have checked with Marsh; he had access to my FB comments.
Throughout his book, Carpenter depends heavily on the opinions of Robert Lindsay (e.g. Chapter 13, p. 184) as expressed in his blog, Beyond Highbrow. Lindsay has been suspended from WordPress.com, and the following screen appears if his link is clicked:
“robertlindsay.wordpress.com is no longer available. This site has been archived or suspended for a violation of our Terms of Service.”
Consequently, we did not argue any of Lindsay’s talking points, because he is disreputable. He’s a polemicist, even to the point of reversing his argument, not a scientist.
Throughout the book (e.g. pp 155-160) Carpenter defends the Ketchum et al. claims that none of the samples were contaminated, that robotics eliminates contamination, that a pristine electropherogram indicates lack of contamination, and that a high Q30 score indicates lack of contamination, all of which are false claims.
Contamination can come from several sources, some outside the control of the analyst:
1. The source is easily contaminated by contact with other animals (e.g. prey, hosts, mates), some of different species (especially microbes). The sloth is said to be a host for 800 different species.
2. A field sample picks up contamination, especially microbial, from the environment before collection. Other animals, including human, may have contacted the sample after it leaves the source.
3. The collector can contaminate the sample, especially if not equipped with forensic equipment.
4. The sample can be contaminated in transport.
5. Lab handlers can contaminate the sample, even with the use of robotics. Robots can be contaminated.
Also, very importantly, use of human specific primers as is done by Family Tree DNA, will only sequence human DNA, which may unknowingly be a contaminant in an otherwise animal sample.
Ketchum disproves her case for no contamination in many ways:
In the video on the SGP webpage (http://sasquatchgenomeproject.org/sasquatch_genome_project_008.htm and
https://www.youtube.com/watch?v=yW4yPPXPfSw) she mentions that her cleaning procedure was to vortex in an alcohol/water mixture. Sykes untrasonicated his hair samples in pure water. DNA is soluble in water, but relatively insoluble in ethanol; hence alcohol it is a poor choice here. Vortexing in water/ethanol may have dislodged some external cells, but would not have dissolved all contaminant DNA, e.g. that not encased in a cell wall. Incidentally, in the same video (at 30.01 minutes) she labels a Sample 140 sequence as cytochrome b, but it actually matches (after translation from negative to positive strand) the control region and HV1, fully 461 base pairs distant. This could just be a reporting error, or it could be a matter of mispriming related to contamination. The 87 bp sequence matches over 100 human accessions in GenBank 100% (87 of 87 bases).
In her video on the SGP webpage, Melba Ketchum only shows pristine electropherograms as evidence for lack of contamination (above). However, on the SGP webpage Supplemental Raw Data tab she also offers for download her electropherograms for S26 as proof. These files require special software for viewing, which I purchased from Heracle Biosoft as DNABaser. Unfortunately, the 272 electropherograms contained only 47 that were good enough to assemble into a sequence of 5260 bp out of the expected 16,568 bp human mitochondrial genome. The remaining 225 electropherograms contained only 11 which could be assembled into a 1923 bp sequence. Thus, 214 of 272 electropherograms were unusable. These showed many overlapping peaks of two or even three different bases, such that sequencing and assembly were impossible.
The first assembly (47 e-grams) aligned with the published S26 mtDNA sequence (Ketchum et al., 2013, Supplemental Data 2), base positions 11401-16551, only 96% with 74 gaps. The second best assembly (11 e-grams) aligned with published S26 base positions 13718 - 15435 only 97% with 27 gaps. This is very poor agreement between this raw data and the published S26 sequence. The 47 e-gram sequence aligned with the 11 e-gram sequence only 93% with 1643 identities and 69 gaps. Thus, only 32% of the S26 mtDNA genome could be sequenced with this data, and the sequence agreed very poorly with the published sequence produced by Family Tree DNA. FTDNA assumes a human sequence and uses human sequencing methodology, so DNA of any other species present would not be detected – it’s what species specific primers do.
Various specific gene sequences from Sample 26 matched human, dog, bear, and unknown, which were probably bacteria. ( See above)
I found that the best hit (most consecutive base pairs) for S31 nDNA was a fungus. The next best hits were all decidedly human. Ketchum acknowledged the "bacterium" to me in a phone call in early October, 2013. At the time I also asked her if she had ever found any DNA evidence for a bear or a dog in her samples. She replied, laughing, “Maybe it’s dogman.” She then asked me not to publish my first paper.
Finally, what is a Q30-score? It is the percentage of base calls that have a less than 1 in 1000 chance of being incorrect. It’s totally based on the electropherogram: peak shapes, peak heights, peak spacing, etc., and has nothing to do with whether the electropherogram is even from the major species in a mixture – that depends on the primers used. So, if human is a trace impurity in a black bear sample, and if only human primers are used, then one gets a human electropherogram in spite of the fact that the major species is black bear. Use black bear primers, and you will get a black bear sequence with no hint of any human impurities. Degraded samples can "self-prime," resulting in extra peaks in the electropherogram," which will overlap the peaks that were from a specific primer. So Carpenter’s and Ketchum’s touting Q30 scores is a red herring: “These scores indicate the purity of the sample.” (p. 157). They are unrelated to sample purity of mixed species samples primed for a single species, unless there are multiple individuals of the same primed species with polymorphic differences, in which case you would get a lower Q30 score based on the SNPs (single nucleotide polymorphisms).
In summary, here are the major fallacies of the Sasquatch Genome Project Study, as propagated by Carpenter in his book, Truth Denied and discussed above:
1. Three whole genomes of the same species were sequenced.
2. These sequences all aligned with one another, confirming a single species.
3. That species is an unknown primate-modern human hybrid, with a mosaic of human and other primate nDNA sequences.
4. The mtDNA was entirely normal human, the result of mating of an unknown primate male with a modern human female.
5. That all the 111 samples were uncontaminated.
“The Study” as reported in (Ketchum et al., 2013) has many other flaws, too numerous to discuss in this book review, but detailed in my blog.
To these Carpenter adds his own fallacies and unproven charges:
1. He accuses Bart Cutino and Tyler Huggins of substituting known black bear samples for alleged S26 samples, which were then sent out for DNA analysis in an attempt to “prove” S26 was a black bear.
2. Dr. Jeff Meldrum and I are accused of claiming to have contacted coauthors of the (Ketchum et al., 2013) paper while not having done so. These additional claims are also false. In fact, as seen above, it is the Ketchum coauthors and supporters who are very closed mouthed about their involvement when challenged with different, supported conclusions.
3. He accuses Nature editors and peer reviewers of colluding with Dr. Bryan Sykes to delay publication of the SGP Study.
4. He maintains (his main theme) that there was a conspiracy among established scientists and some amateur bigfooters alike to suppress the Sasquatch Genome Study and to dishonor Dr. Melba Ketchum and her reputation for having challenged conventional wisdom. But he produces no hard evidence, just speculation. Criticism of the paper was not organized, rather independent opinions by various individuals, including me. Some were scientists, some were not. For example, I received no assistance and very minimal encouragement (all in retrospect) for my work.
5. He charges that the peer review process is designed to preserve the status quo and prevent revolutionary new discoveries from seeing the light of day. Reputable journals have very strict rules governing peer reviews, designed to be fair to all.
6. He implies that somehow the discovery of ancient hominin fossils around the world validates the Ketchum et al. Study and negates its specific criticisms (a theme also prevalent on Melba Ketchum’s FB page). The above specific scientific criticisms of the Ketchum et al.(2013 paper) touch only on the samples, analyses, results, and conclusions discussed therein, none of which are connected to other discoveries of ancient hominins. Criticisms above do not involve the larger question of the existence of sasquatch either.
7. He claims that independent studies support the findings of the Sasquatch Genome Study (Chapter 15, p. 198), but then refers to irrelevant ancient hominin studies, Go Fund Me supporter comments, inconclusive purported orang pendek results, and armchair statements by supporters, none of whom have ever published their results in detail in over six years since the Ketchum et al. (2013) paper was published. There have been no published (let alone peer reviewed) scientific studies validating the Ketchum et al. Study that I am aware of. There are at least two published, peer reviewed rebuttals (Hart, 2016a, 2016b).
Carpenter is an advocate of citizen science, and so are we. Activities like collecting field samples, footprint casting, and audio and video recording are one thing, but rechecking DNA sequence matches and associated calculations, interpreting light and electron micrographs, and judging whether the method of DNA analysis is appropriate for the sample and the study objectives involves a much higher level of scientific education, background, and understanding. Carpenter shows no evidence of this or even that he has ever personally done a simple BLAST® search (free online through NCBI), which should be a rather simple matter for a BS in computer science and systems administrator.
Consequently, Truth Denied is not a scientifically based review of the Ketchum et al. Study and its criticisms. It’s a story of alleged conspiracy, intrigue, malfeasance, and character assassination. Conspiracies are easy to charge, but almost always difficult to prove, otherwise they would be called news or history. The real conspiracy here is the conspiracy of silence among Ketchum coauthors, some sample submitters, and others associated with the SGP, enforced through NDAs. Real science does not operate on NDAs once a paper is published. Open dialogue is a key element in its advancement. If you like mysteries, this book may be for you. If you seek open, honest, competent, scientific debate on the complex technical subject of DNA analysis of these purported sasquatch samples, look elsewhere or see above.
Haskell V. Hart
Canyon Lake, Texas, USA
December 6, 2019
Note: This reviewer saw a sasquatch in broad daylight in the Ouachita Mountains of southeast Oklahoma on June 6, 2018, at 11:20 AM.
Bibliography
Altschul SF et al. (1990) Basic local alignment search tool. Journal of Molecular Biology 215 (No. 3): 403-410. https://blast.ncbi.nlm.nih.gov/Blast.cgi
Hart HV (2016a) Not finding bigfoot in DNA. Journal of Cryptozoology 4: 39-51.
Hart HV (2016b) DNA as evidence for the existence of relict hominoids. 5: 8-31. https://www.isu.edu/media/libraries/rhi/research-papers/HART-DNA-Evidence.pdf
Ketchum M. S. et al. (2013) Novel North American hominins: next generation sequencing of three whole genomes and associated studies. DeNovo 1:1. Online only: http://sasquatchgenomeproject.org/sasquatch_genome_project_002.htm
Linacre AMT and Tobe SS (2013) Wildlife DNA Analysis: Applications in Forensic Science. : Chichester (UK): Wiley-Blackwell.
Meldrum J (2006) Sasquatch: Legend Meets Science. New York: Tom A. Doherty Associates.
Meldrum J (2013) Sasquatch Field Guide: Identifying, Tracking, and Sighting North America’s Relict Hominoid. Arcata: Paradise Cay Publications.
Sykes BC et al. (2014). Genetic analysis of hair samples attributed to yeti, bigfoot and other anomalous primates. Proceedings of the Royal Society B 281: 20140161. http://rspb.royalsocietypublishing.org/content/281/1789/20140161.